Back to culture introduction
Back to culture introductionOriginally written in May 2002 with 2008 updates about the cysteine requirement and cloning procyclic forms(also available in pdf) © George A.M. Cross
Back to culture introduction![]() Home
Home
Bloodstream forms (BF)
There are some records of early attempts to maintain BF Trypanosoma brucei in vitro [1, 2], but short-term propagation, in the presence of feeder cells (L-cells), was not achieved until 1967 [3]. Continuous propagation was not reported until 1977, when the Lister 427 strain (clone 052 from GAMC) was successfully propagated in RPMI 1640 medium with 25 mM HEPES and 20% heat-inactivated FBS, in the presence of bovine primary fibroblast-like feeder cells [4-6]. This successful result was obtained in an experiment in which 96 permutations of 12 different media, with varying amounts of FBS, ± feeder cells, were tested simultaneously. It initially only worked for the Lister 427 strain.
Brun and colleagues [7, 8] introduced MEM (Minimal Essential Medium of Eagle) supplemented with MEM non-essential amino acids, 30 mM HEPES pH 7.5, and glutamine, and experimented with feeder cells and sera, to find conditions that allowed any T. brucei stock to be cultivated. Comparing embryonic fibroblast-like lines from rabbits and a north American vole, Microtus montanus, the latter were found to be better (and we also used these in the early 1980s). Heat-inactivated 15% human or fresh (but not commercially supplied) rabbit serum supported much better growth than FBS.
An accidental observation by Michael Duszenko, in our laboratory, led to the rational elimination of feeder cells from the MEM-based culture system [9], and the demonstration that cysteine is an essential growth requirement but is actually toxic to trypanosomes at mM concentrations. After preparing MEM from individual ingredients, for reasons that are not relevant here, and observing that the trypanosomes died immediately upon being placed in the freshly dissolved medium, Duszenko recognized that he had made an error by adding cysteine instead of cystine. A literature search, using the search terms 'CYSTEINE & CELL & CULTURE' identified three studies, published from 1975-1982, which demonstrated, in a mammalian culture system, that feeder cells can take up cystine and excrete cysteine, which was otherwise toxic to the cells being cultured. We were immediately able to demonstrate that the repeated addition of sub-toxic concentrations of cysteine eliminated the feeder-cell requirement for T. brucei culture. We then showed that, by adding more stable thio compounds, we could buffer the cystine-cysteine equilibrium, eliminating the need for continuous low-level infusion of cysteine. Another laboratory made a similar observation empirically, without identifying the specific explanation, during a broad survey of conditions affecting trypanosome culture [10]. They concluded, in fact, that "the probable role of the reducing agents [they included mercaptoethanol or thioglycerol in their media] is to stabilize different essential components of the medium, such as vitamins and glutathione". They proposed that reduced glutathione might be important to protect trypanosomes against activated oxygen species, since Meshnick had shown, in 1978, that T. brucei lacked catalase and glutathione reductase. Duszenko's subsequent experiments showed that trypanosomes took up cysteine, but not cystine, and proved that it was an essential amino acid, not just a reducing factor [11].
What we had not noted at this time is that cysteine 'autoxidation' is actually catalyzed by copper. In 1989, Hirumi & Hirumi published the HMI-9 medium [12], based on Iscove's modification (IMDM) of Dulbecco's MEM (Modified Eagles Medium), that is now most widely used for culture of BF T. brucei, and introduced a copper chelator, bathocuproine sulfonate, whose use for the culture of Pneumocystis carinii, in the absence of feeder cells, had been published the previous year. Bathocuproine, at 0.05 mM, extended the life of the 1.5 mM cysteine-containing medium for at least 10 days (the maximum shelf life was not established). Addition of 2 mM pyruvate was also required to support the growth of BF during primary cultivation, but not once the cultures were established. This paper also reported growth in a serum-free version of HMI-9 (HMI-18), in which the Serum Plus™ component was increased from 10% to 20%. Serum Plus&#; contains 20% serum (JRH Biosciences, personal communication).
Either supplemented MEM (used by the Duszenko & Brun laboratories), or IMDM-based HMI-9, seem equally suitable for BF culture. As is true for many cells, a short period of adaptation, assisted by transitional subcultures in mixed media, is helpful when switching culture media.
In 1992, when we resurrected BF culture in our laboratory, to initiate transfection experiments, we unexpectedly found that BF grew extremely well, with high plating efficiency, on agarose plates containing HMI-9 medium [13]. The cell density in the colonies was close to 109/ml (106 per colony), which seemed quite amazing. Why can the cells grow to this density in rodents and on plates, but only to about 2x106/ml in liquid culture? Although this paper also reported growth of pleomorphic strains on agarose, and issues of adaptation to liquid or agarose culture, these observations were apparently overlooked in a subsequent and more extensive analysis of the growth of pleomorphic strains [14].
What are 'pleomorphic' strains and why are they more difficult to grow in culture? Pleomorphic strains are those that, by definition, show varied morphology in their BF, ranging from slender, through 'intermediate' to 'stumpy' forms. Pleomorphism is generally associated with 'recent isolates' that have not been adapted to virulent growth in laboratory rodents, but this is not exclusively so. Pleomorphic isolates do not exhibit high parasitemias in mice and achieve even lower densities in culture than the prototypical Lister 427 strain. The key reasons are that only the slender forms divide---stumpy forms are non-dividing forms that are primed to differentiate to PF, in the tsetse---and that some kind of quorum-sensing mechanism exists by which trypanosomes apparently limit their growth density, both in vivo and in culture. BF trypanosomes secrete an unidentified low molecular mass substance, dubbed Stumpy Induction Factor (SIF), which induces cell cycle arrest, manifested as differentiation of slender to stumpy forms through a cAMP-mediated pathway [15-17]. The challenge of identifying SIF has eluded several investigators.
Even the virulent Lister 427 strain does not produce the yield that might be anticipated, given the rich environment provided by the culture media. Cultures enter stationary phase at around 2x106/ml, and produce stumpy-like forms (see below), so the yield limitation is probably also attributable to SIF. The more virulent strains are good producers of SIF but are apparently less susceptible to its effects.
Propagation of T. brucei BF in a medium (HMI-244) completely lacking serum components, has been reported [18]. This study used one strain each of T. brucei rhodesiense and T. evansi, so direct comparison with T. brucei Lister 427 in HMI-9 is not possible. The maximum cell densities for each strain were ~2x106/ml, and the population doubling time was ~11 hours. As lower growth rates were obtained with seeding densities below 5x104 per ml, serum-free media are unsuitable for cloning. The concentrations of some supplements were very critical, and the media may be deficient in some requirements (fatty acids and lipids, for example).
Lister 427 BF can be cloned without any special care, in HMI-9 liquid medium or on agar plates. In contrast to PF (see below), BF do not appear to have any minimum density requirement.
Procyclic (tsetse midgut) forms (PF)The term 'procyclic' was coined by John Baker [19]. Until 1970, trypanosomes isolated from infected animals or humans could only be propagated on 'blood-agar' media, generally containing fresh defibrinated rabbit blood, at ~27°C, in a non-infective form that corresponded, morphologically and physiologically, to the tsetse midgut stage of the life cycle. The trypanosomes divided in the small layer of fluid lying over the solid blood-agar 'slopes'.
A significant improvement in procyclic culture was reported in 1970 (M.D. Pittam: appendix to [20]). This medium consisted of a phosphate-buffered lysate of settled but unwashed human red blood cells. We and others attempted to use Pittam's 'monophasic culture medium' (MCM) to study mitochondrial biogenesis of T. brucei during transformation from BF to PF. Although the use of fresh defibrinated horse blood provided the most reproducible growth (reported in [21], along with detailed instructions for preparing MCM), the variation in biochemical assays led me to drop this project, pending the development of a defined culture medium [21]. This is another old paper that is overlooked, which reported a chemically defined medium (HX25) in which PF could be continuously propagated in the absence of serum, and even in the absence of any protein! One feature of this medium is its high osmolarity. A few modifications to HX25 (HX25M) and a related medium (HX28) are described in a later publication [22], which focussed on the amino acid consumption of PF T. brucei and identified threonine as a major source of acetate.
Part of the discussion from [21] is reproduced below.
The yield of organisms in both media HXl2V and HX25 is not as high as one might hope to achieve in such ostensibly nutritious environments, and this suggests that some unidentified factor may be present at growth-limiting concentration. We have not explored the important area of trace metal requirements and further studies in this direction might be desirable. Alternatively, it may be the accumulation of a toxic metabolite which limits growth in our media.
The defined medium described is not a minimal medium: no attempt has yet been made to simplify its composition.
It is possible that the success of HX25, in which we could propagate the three strains that we tested, stimulated others to experiment with synthetic media. SDM-79 [23], which is widely used and based on the same Medium 199 formulation used in HX25. Its original formulation was quite complex, and has been superseded by a simpler overall formulation, which is available commercially. The MEM formulation used by the Duszenko lab for BF culture (see above) can be used for differentiation to PF (see below) when supplemented with proline (600 mg/l), hemin ( 7.5 mg/l), citrate (882 mg/l) and cis-aconitate (522 mg/l). PF can be maintained in the same medium without citrate, cis-aconitate, cysteine, bathocuproine, or supplemental (to MEM) glucose. In principal using almost the same medium for BF and PF should simplify laboratory management, but I do not know if MEM gives the same high yield of cells as do Cunningham's and SDM-79.
Another medium, with a very different composition, was designed by Isabel Cunningham [24]. It is based on Grace's insect-cell culture medium, modified to more closely mimic the amino acid composition of tsetse hemolymph, but conspicuously lacking purines, which trypanosomes and several other parasitic protozoa cannot synthesize de novo. It is used in several laboratories. We have even experimented with mixtures of SDM-79 and Cunningham's medium. The MEM-based modified DTM (see below in the section on bloodstream to procyclic differentiation) is also suitable for the continued propagation of procyclic forms. SDM-79, Cunningham's, and DTM all contain serum.
In the original version of this commentary (2nd May 2002), I wrote that “Cloning PF is frustrating. Something needs to be done about this! If the culture density is allowed to drop below around 104 cells per ml, the cultures die. For cloning PF, many investigators use ‘conditioned medium’, which means a 50% mix of fresh medium with 50% of the medium in which PF have grown to about 107/ml. Conditioned medium increases plating efficiencies on agar [13]. Some investigators, when cloning transfected PF, add 105 non-transfected cells per ml, but others find that a 5% CO2 gas phase obviates the need for conditioned medium (in medium containing sodium bicarbonate, presumably, to allow CO2 buffering). So far, no-one has checked all four variations of these two parameters simultaneously, to my knowledge. There may be a very simple explanation and solution to the density issue: gas phase, oxygen reduction, scavenging of free radicals, etc. Perhaps adding cysteine and bathocuproine, as for BF, would solve the problem!”
Since 2004, however, we have taken the advice of Paul Englund’s lab, which has been reiterated for almost as long on our web site, that raising the serum concentration from 10% to 15% results in a “drastic improvement in cloning efficiency” (quoting Paul Englund); ~50% in our hands. When updating this commentary in 2008, I asked Paul if this had been published. The answer was yes; in July 2002 (Klingbeil, M. M., S. A. Motyka, and P. T. Englund. 2002. Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol Cell 10:175-186 DOI PMID), which says they were “…cultured in SDM-79 medium containing 15% fetal bovine serum ... cloned by limiting dilution in … 96-well tissue culture plates at 1 cell/ml under 5% CO2 at 27°C (cloning method from W. Gibson, University of Bristol) … plating efficiencies ranging from 30%–60%”.
Differentiation of BF to PFThe most influential proponent of the relationship between morphology, mitochondrial physiology, and life-cycle transitions of T. brucei, in the period from 1960-1980, was Keith Vickerman. His 1965 review [25] is essential reading, and he published other seminal studies on trypanosome morphology and life-cycle changes. His writing is exemplary and persuasive, and his papers are thoroughly documented with references to the prior art. Early indications of major changes in oxidative metabolism during the life cycle are attributable to the biochemical studies of John Ryley and colleagues, during the 1950s and 1960s.
BF differentiates quite readily to PF in vitro. This is what the first cultures, in the early 20th century, represented. It was known for many years that a predominance of stumpy forms led to easier differentiation in culture, and establishment of tsetse infections. In recent years, this has been validated by several labs and it is generally accepted that cells in G0 are prepared to differentiate and simultaneously resume the cell cycle [26]. Although Lister 427 is predominantly monomorphic [27] and does not noticeably produce G0-arrested stumpy forms in healthy cultures or in acute animal infections, it can nevertheless differentiate efficiently, but not always as synchronously as is the case with a strain from which a homogeneous population of stumpy forms can be obtained. One can see stumpy forms in relapsing infections (Lister 427 usually undergoes only one relapse in mice or rats, because it is so virulent: rabbits allow long-term infection modeling, but parasitemias are very low), but there are two published reports of differentiation from slender to stumpy forms in culture [28, 15]. Stumpy forms can be seen in stationary-phase BF cultures, and occurred more readily with variant 118, rather than the commonly used clones that express VSG 221 (David Horn, personal communication, 2001). The general debate about monomorphic and pleomorphic BF lines, and the reversal of monomorphism, are beyond the intended scope of this document, but are discussed elsewhere.
Efficient differentiation of pleomorphic BF to PF in SDM-79 medium was originally shown to require reducing the temperature from 37°C to 27°C and adding ≥ 3 mM citrate and/or cis-aconitate (CCA) [29]. These compounds were tested on the idea that they might prime the TCA cycle, which becomes active in the PF, but we still have no satisfactory explanation of the role of cis-aconitate, despite additional studies by several laboratories (for examples, see [30-33]). A more recent study showed that lowering the temperature to 20°C reduced to micromolar levels the cis-aconitate or citrate concentration required for efficient differentiation of pleomorphic stumpy but not slender BF [48].
The most widely used differentiation protocols are based on DTM, Differentiating Trypanosome Medium [32]. This medium was identical to the TM (Trypanosome Medium, based on MEM) used for BF culture (in the presence of 15% fresh heat-inactivated horse serum and feeder cells), except that it lacked glucose and was supplemented with proline. In these experiments [32], in which the monomorphic Lister 427 clones 221 and 117 were compared with the pleomorphic AnTat 1.1 strain, BF were obtained from mice or from cultures on Microtus feeder cells. This study confirmed that both CCA and a temperature drop were required for differentiation. BF died after 48 h in the presence of CCA when maintained at 37°C, at which time there was already a strong induction of TCA-cycle enzymes characteristic of PF. After 17 h in CCA at 37°C, many cells in the population resembled stumpy forms. A few cells that escaped death from CCA at 37° apparently adapted to the situation and continued to divide as BF: "…in summary, it appears that at 37°C in the presence of CCA the cells have the choice of either undergoing an abortive differentiation to non-proliferating procyclic-like cells or retaining the properties of dividing bloodstream forms". In these experiments, Lister 427 clones appeared to differentiate and divide without any lag. Differentiation of stumpy forms of the pleomorphic AnTat 1.1 still required CCA, so this is not a peculiar requirement for differentiation of monomorphic lines. As judged by loss of VSG and appearance of several PF-markers, including procyclin, BF can also be triggered to differentiate into PF, and continue to divide at 37°C, if glucose is removed from an MEM-based BF culture medium and replaced by glycerol [34]. Although this transformation was less efficient than when induced by CCA and temperature shift (the two conditions were not directly compared, but loss of VSG and appearance of procyclin were far slower than in published CCA/temperature shift experiments), the authors concluded that "…the independence of this [transformation] from these 'essential' in vitro triggers may be [physiologically] significant". These investigators also noted that their BF cultures (Lister 427 clone 117) displayed some features suggestive of partial transition to PF-type metabolism (being partially resistant to SHAM/glycerol treatment), and that the PF arising could have represented a small part of the initial BF population, most of which died during the induction procedure. These authors mention a 1972 abstract reporting the continuous propagation of PF at 37°C, which was also reported to me in the 1980s (Lex van der Ploeg, personal communication), but never tried in our lab.
Treating BF with DFMO (Eflornithine), which depletes intracellular putrescine, arrests cell division and induces stumpy morphology [35, 36]. DFMO-treatment of an otherwise monomorphic cell line was reported to allow synchronous transformation to PF, when subsequently subjected to CCA and temperature drop triggers [37]. However, no specific markers of differentiation, other than morphology and division, were used in this study. There are several other studies of differentiation, but it is difficult to make meaningful comparisons of some studies, which were often performed with different trypanosome strains using different media for growth and differentiation. Altogether, we have not moved beyond CCA/temperature as the most effective triggers, nor to understanding why they are.
In a later publication [38] (and in the formulation, with Overath's handwritten notes, provided to GAMC in April 1994), DTM was further modified by the addition of glycerol (in place of glucose), heme, 15% fetal bovine serum, 0.2 mM 2-mercaptoethanol, and extra proline, glutamate, glutamine. There is an error in the published [38] formulation: glycine is stated where glycerol was intended. Vassella and Boshart further modified DTM by adding 28.2 mg/l bathocuproine and 182 mg/l cysteine [14].
The kinetics of VSG loss and procyclin appearance, during differentiation under the same conditions, have been described in the 117 clone of Lister 427 [39], and there have been many additional studies of differentiation of this and other trypanosomes strains, whose review is beyond the scope of this document. Differentiation of pleomorphic strains is described in several studies [38, 26, 17, 40].
The protocol generally used in our lab for differentiation of Lister 427, was documented by David Horn, as follows. 107log-phase BCF are washed in 5 ml DTM at 30°C then resuspended in 5 ml DTM + 3 mM CCA at 27°C. Cell density is maintained, by serial subculture, between 1 and 6 x 106/ml. In one of several examples of 'protocol drift', I am informed that the DTM wash is not currently used in my lab!
Oddly, and for no identified reason, recently differentiated PF do not grow, in our hands, to the same high densities (5x107/ml) as longer established PF. They also have a lower survival rate when electroporated---more like BF than established PF.
Differentiation of procyclics to infectious pseudo-metacyclic or bloodstream formsThere have been sporadic reports of the requisition of infectivity by PF trypanosomes from blood-agar cultures [41, 42]. Some investigators pursued the idea of trying to culture trypanosomes in the presence of tsetse salivary-gland tissue and organ explants [24, 43-46]. I have reported conditions under which a substantial proportion of non-dividing PF cells, previously maintained for long periods as non-infective PF, reacquired the ability to infect mice. The factors that led to this, apart from cessation of growth (perhaps they were fortuitously arrested in G0, as for the efficient reverse differentiation from BF to PF---but remember the proliferating epimastigote form intervenes, in vivo, between the procyclic and metacyclic forms), were never determined. Variant clone 060 (MITat 1.1), of Lister 427 was derived from these infective forms. The most apparently reproducible technique for generating metacyclic-like forms [47] has been replicated by others (Wendy Gibson, personal communication, 1994) and works with Lister 427. One concern in this work, which is addressed in the discussion of the paper, but not completely satisfactorily, is whether any of the infective forms in these cultures were simply surviving or incompletely differentiated BF. The method was not tested with long-term established PF, and there is no obvious reason why metacyclic forms appeared under conditions of continuous culture. Unless the method works with long-term PF cultures, its use for genetic studies would be somewhat limited, but it could be interesting for studies of metacyclic VSG expression.
Why is it so difficult to differentiate from procyclics back to bloodstream forms? Are there any logical things that could be tried to stimulate this transformation? There are anecdotal reports of adding cAMP or its permeable derivatives to procyclic cultures, without triggering differentiation. Could the change be stimulated by depletion of heme? In our experiments, were the procyclic forms fortuitously arrested in G0, and thereby rendered infectious? Some imaginative new studies are needed in this area.
An update on the Cysteine requirement
A poster — Analysis of cysteine biosynthesis in Leishmania by McCraig, Silva, Williams, Coombs & Muller — at the BSP meeting in Newcastle in March 2008 prompted me to look for the genes for cysteine biosynthesis in the trypanosome genomes. In their abstract, the authors noted that “Leishmania acquire cysteine through two distinct pathways — trans-sulfuration and de novo biosynthesis”.
De novo synthesis involves two steps: serine O-acetylation by serine acetyl transferase (EC 2.3.1.30) then addition of sulfur by cysteine synthase (EC 2.5.1.47). The prototypical source of sulfur is given as H2S, but where would kinetoplastids get this? The trans-sulfuration pathway is for the interconversion of homocysteine and cysteine via cystathionine.
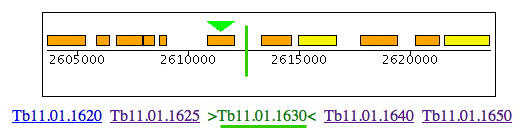
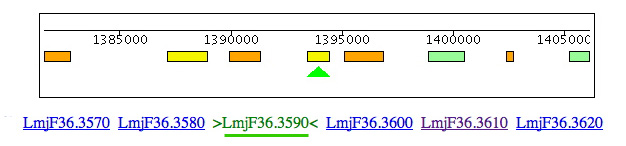
Leishmania and Trypanosoma cruzi contain genes for cysteine synthase (Lmjf36.3.3590 & Tc00.1047053507165.50 & Tc00.1047053507793.20) but, in the region of synteny between Leishmania chromosome 3 and T. brucei chromosome 11, the cysteine synthase is clearly absent from T. brucei but 4 of 5 of the adjacent genes (LmjF36.3620/ Tb11.01.1625, LmjF36.3610/Tb11.01.missing, LmjF36.3600/Tb11.01.1630, LmjF36.3590/Tb11.01.missing, LmjF36.3580/Tb11.01.1640 & LmjF36.3570/Tb11.01.1650) are conserved. The distance between Tb110.0.1630 & 1640 is only 1219 bp. A BLAST search of Lm with this DNA sequence shows no significant match, so the gene seems to be truly absent from T. brucei.
 |
|---|
|
|
|
The absence of cysteine synthase from T. brucei could be an explanation for the famous requirement of T. brucei for exogenous cysteine, but this also raises the question of why are we providing cysteine for BF but not for PF?
All three species have cystathionine synthase (EC 4.2.1.22) (LmjF17.0250 & Tc00.1047053508177.120 are syntenic & Tb11.02.5400 is not) which has highly similarity (e-39) with cysteine synthase, to convert homocysteine to cystathionine, and cystathionine lyase (EC 4.4.1.1) (Tb09.211.3330, LmjF35.3230, Tc00.1047053510661.250) to convert cystathionine to cysteine, but T. brucei media do not contain homocysteine and it too, presumably, would be susceptible to copper-catalyzed oxidation in the absence of bathocuproine.
Incidentally, some people have asked if I know why Taurine is present (and at high concentration) in SDM79 and Cunningham’s medium. I have no idea, but I am sure it is unnecessary. Taurine, in which the sulfur is oxidized, can be a product of cysteine degradation but does not seem to be a useful source of anything.
References
[1] Yorke W, Adams ARD, Murgatroyd F. Ann Trop Med Parasitol 1929;23:
[2] Williamson J, Rollo IM. Stimulating effect of amino acids on the survival at 37°C of Trypanosoma rhodesiense in a serum-free synthetic medium. Nature 1952;170:376-7.
[3] Le Page RW. Short term cultivation of Trypanosoma brucei in vitro at 37 degrees C. Nature 1967;216:1141-2.
[4] Hirumi H, Doyle JJ, Hirumi K. African trypanosomes: cultivation of animal-infective Trypanosoma brucei in vitro. Science 1977;196:992-4.
[5] Doyle JJ, Hirumi H, Hirumi K, Lupton EN, Cross GAM. Antigenic variation in clones of animal-infective Trypanosoma brucei derived and maintained in vitro. Parasitology 1980;80:359-69.
[6] Hirumi H, Hirumi K, Doyle JJ, Cross GAM. In vitro cloning of animal-infective bloodstream forms of Trypanosoma brucei. Parasitology 1980;80:371-82.
[7] Brun R, Jenni L, Tanner M, Schonenberger M, Schell KF. Cultivation of vertebrate infective forms derived from metacyclic forms of pleomorphic Trypanosoma brucei stocks. Acta Trop 1979;36:387-90.
[8] Brun R, Jenni L, Schonenberger M, Schell KF. In vitro cultivation of bloodstream forms of Trypanosoma brucei, T. rhodesiense, and T. gambiense. J Protozool 1981;28:470-9.
[9] Duszenko M, Ferguson MAJ, Lamont GS, Rifkin MR, Cross GAM. Cysteine eliminates the feeder cell requirement for cultivation of Trypanosoma brucei bloodstream forms in vitro. J Exp Med 1985;162:1256-63.
[10] Baltz T, Baltz D, Giroud C, Crockett J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J 1985;4:1273-7.
[11] Duszenko M, Muhlstadt K, Broder A. Cysteine is an essential growth factor for Trypanosoma brucei bloodstream forms. Mol Biochem Parasitol 1992;50:269-74.
[12] Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei bloodstream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 1989;75:985-9.
[13] Carruthers VB, Cross GAM. High-efficiency clonal growth of bloodstream- and insect-form Trypanosoma brucei on agarose plates. Proc Natl Acad Sci USA 1992;89:8818-21.
[14] Vassella E, Boshart M. High molecular mass agarose matrix supports growth of bloodstream forms of pleomorphic Trypanosoma brucei strains in axenic culture. Mol Biochem Parasitol 1996;82:91-105.
[15] Hesse F, Selzer PM, Muhlstadt K, Duszenko M. A novel cultivation technique for long-term maintenance of bloodstream form trypanosomes in vitro. Mol Biochem Parasitol 1995;70:157-66.
[16] Reuner B, Vassella E, Yutzy B, Boshart M. Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol Biochem Parasitol 1997;90:269-80.
[17] Vassella E, Reuner B, Yutzy B, Boshart M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J Cell Sci 1997;110:2661-71.
[18] Hirumi H, Martin S, Hirumi K, Inoue N, Kanbara H, Saito A, Suzuki N. Cultivation of bloodstream forms of Trypanosoma brucei and T. evansi in a serum-free medium. Tropical Medicine & International Health 1997;2:240-4.
[19] Newton BA, Cross GAM, Baker JR. (1973) Differentiation in Trypanosomatidae. in: Symposia of the Society for General Microbiology: Microbial Differentiation, 23 (Eds J. E. Smithand J. M. Ashworth, Cambridge University Press, Cambridge) p. 339-73.
[20] Dixon H, Williamson J. The lipid composition of blood and culture forms of Trypanosoma lewisi and Trypanosoma rhodesiense compared with that of their environment. Comp Biochem Physiol 1970;33:111-28.
[21] Cross GAM, Manning JC. Cultivation of Trypanosoma brucei sspp. in semi-defined and defined media. Parasitology 1973;67:315-31.
[22] Cross GAM, Klein RA, Linstead DJ. Utilisation of amino acids by Trypanosoma brucei in culture: L-threonine as a precursor for acetate. Parasitology 1975;71 :311-26.
[23] Brun R, Schonenberger M. Cultivation and in vitro cloning of procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop 1979;36:289-92.
[24] Cunningham I. New culture medium for maintenance of Tsetse tissues and growth of trypanosomes. J Protozool 1977;24:325-9.
[25] Vickerman K. Polymorphism and mitochondrial activity in sleeping sickness trypanosomes. Nature 1965;208:762-6.
[26] Matthews KR, Gull K. Commitment to differentiation and cell cycle re-entry are coincident but separable events in the transformation of African trypanosomes from their bloodstream to their insect form. J Cell Sci 1997;110:2609-18.
[27] Bohringer S, Hecker H. Quantitative ultrastructural differences between strains of the Tryponasoma brucei subgroup during transformation in blood. J Protozool 1974;21:694-8.
[28] Hamm B, Schindler A, Mencke D, Duszenko M. Differentiation of Trypanosoma brucei bloodstream trypomastigotes from long slender to short stumpy-like forms in axenic culture. Mol Biochem Parasitol 1990;40:13-22.
[29] Brun R, Schonenberger M. Stimulating effect of citrate and cis-aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z Parasitenkd 1981;66:17-24.
[30] Overath P, Czichos J, Stock U, Nonnengaesser C. Repression of glycoprotein synthesis and release of surface coat during transformation of Trypanosoma brucei. EMBO J 1983;2:1721-8.
[31] Czichos J, Nonnengaesser C, Overath P. Trypanosoma brucei: cis-aconitate and temperature reduction as triggers of synchronous transformation of bloodstream to procyclic trypomastigotes in vitro. Exp Parasitol 1986;62:283-91.
[32] Overath P, Czichos J, Haas C. The effect of citrate/cis-aconitate on oxidative metabolism during transformation of Trypanosoma brucei. Eur J Biochem 1986;160:175-82.
[33] Saas J, Ziegelbauer K, von Haeseler A, Fast B, Boshart M. A developmentally regulated aconitase related to iron-regulatory protein-1 is localized in the cytoplasm and in the mitochondrion of Trypanosoma brucei. J Biol Chem 2000;275:2745-55.
[34] Milne KG, Prescott AR, Ferguson MAJ. Transformation of monomorphic Trypanosoma brucei bloodstream form trypomastigotes into procyclic forms at 37°C by removing glucose from the culture medium. Mol Biochem Parasitol 1998;94:99-112.
[35] Bacchi CJ, Garofalo J, Mockenhaupt D, McCann PP, Diekema KA, Pegg AE, Nathan HC, Mullaney EA, Chunosoff L, Sjoerdsma A, Hutner SH. In vivo effects of alpha-DL-difluoromethylornithine on the metabolism and morphology of Trypanosoma brucei brucei. Mol Biochem Parasitol 1983;7:209-25.
[36] deGee ALW, Carstens PHB, McCann PP, Mansfield J. Morphological changes in Trypanosoma brucei rhodesiense folowing inhibition of polyamine biosynthesis in vivo. Tissue and Cell 1984;16:731-8.
[37] Giffin BF, McCann PP. Altered intracellular polyamines in bloodstream form Trypanosoma brucei brucei: transformation to procyclic trypomastigotes. Acta Trop 1993;55:181-90.
[38] Ziegelbauer K, Quinten M, Schwarz H, Pearson TW, Overath P. Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro. Eur J Biochem 1990;192:373-8.
[39] Roditi I, Schwarz H, Pearson TW, Beecroft RP, Liu MK, Williams RO, Overath P. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J Cell Biol 1989;108:737-46.
[40] Matthews KR. Developments in the differentiation of Trypanosoma brucei. Parasitol Today 1999;15:76-80.
[41] Trager W. Tsetse-fly tissue culture and the development of trypanosomes to the infective stage. Annals Trop Med Parasitol 1959;53:473-91.
[42] Amrein YU, Geigy R, Kauffmann M. On the reaquisition of virulence in trypanosomes of the brucei group. Acta Tropica 1965;22:193-203.
[43] Cunningham I, Honigberg BM, Taylor AM. Infectivity of monomorphic and pleomorphic Trypanosoma brucei stocks cultivated at 28 °C with various tsetse fly tissues. JParasitol 1981;67:391-7.
[44] Cunningham I. Infectivity of Trypanosoma rhodesiense cultivated at 28°C with various tsetse fly tissues. J Protozool 1986;33:226-31.
[45] Kaminsky R, Beaudoin E, Cunningham I. Studies on the development of metacyclic Trypanosoma brucei sspp. cultivated at 27°C with insect cell lines. J Protozool 1987;34:372-7.
[46] Kaminsky R, Beaudoin E, Cunningham J. Cultivation of the life cycle stages of Trypanosoma brucei sspp. Acta Trop 1988;45:33-43.
[47] Hirumi H, Hirumi K, Moloo SK, Shaw MK. Trypanosoma brucei brucei: in vitro production of metacyclic forms. J Protozool 1992;39:619-27.
[48] Engstler M, Boshart M. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev 2004;18:2798-811.